

| Cellular and Molecular Medicine Research, ISSN 2817-6359 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cell Mol Med Res and Elmer Press Inc |
| Journal website https://www.thecmmr.org |
Review
Volume 1, Number 1, September 2023, pages 12-19

Recent Advances in Characterization of Pre-Leukemic/Leukemic Stem Cells in Acute Myeloid Leukemia
Liqing Jina, b, Licun Wua
aPrincess Margaret Cancer Center, University Health Network, Toronto, Ontario M5G 1L7, Canada
bCorresponding Author: Liqing Jin, Princess Margaret Cancer Center, University Health Network, 101 College St., Toronto, Ontario M5G 1L7, Canada
Manuscript submitted June 8, 2023, accepted July 3, 2023, published online August 30, 2023
Short title: Pre-Leukemic/Leukemic Stem Cells in AML
doi: https://doi.org/10.14740/cmmr18e
- Abstract
- Introduction
- Characteristics of LSCs
- Clinical Relevance of LSCs
- Pre-Leukemic HSCs in AML
- Cell Origin of AML Relapse
- Conclusions
- References
| Abstract | ▴Top |
Acute myeloid leukemia (AML) is a heterogeneous disease organized as a hierarchy similar to normal hematopoiesis. It is maintained and progresses by self-renewing leukemia stem cells (LSCs) that are usually quiescent and resistant to current chemotherapies and thus, contribute to leukemia relapse. Recently, more ancestral pre-leukemic hematopoietic stem cells (HSCs) were identified, suggesting that accumulation of mutations occurs in self-renewing HSCs. The pre-leukemic/leukemic stem cell model proposes that a non-genetic mechanism in leukemic process, harmonized together with the genetic model and recently identified epigenetic dysregulations, contributes to leukemia heterogeneity and treatment resistance. Here we review the recent advances in characterization of AML LSCs, identification of pre-leukemic HSCs, and emerging concept of cell origin that contributes to AML relapse. These profound insights into AML biology have clinical importance and great implications for future development of new anti-AML therapies.
Keywords: Acute myeloid leukemia; Leukemia stem cell; Pre-leukemic hematopoietic stem cell
| Introduction | ▴Top |
Acute myeloid leukemia (AML) is a biologically, molecularly, and clinically heterogeneous disease with variant clinical outcomes. Despite advances in treatment over recent decades, except acute promyelocytic leukemia (APL, M3 by FAB classification), sustained remission and long-term survival have not dramatically improved with more than 70% of AML patients dying of disease relapse in less than 5 years. Development of new therapies depends on our understanding of AML biology and genetics. For example, fundamental discoveries of chromosomal translocation and molecular mutations (e.g. Flt3, NPM1 and epigenetic modifiers DNMT3a, TET2, IDH1/2) in AML not only allow clinicians subgroup patients with favorable and adverse outcomes for therapeutic options and choices following induction treatment, but also provide opportunities for therapeutic targeting [1]. In parallel to the continuous effort to investigate the genetic abnormalities for understanding the pathophysiology of AML, the nature and existence of leukemia stem cells (LSCs) in leukemia were also identified and extensively characterized in last two decades [2, 3]. LSCs sit at the apex of leukemia to maintain leukemia clones and contribute to leukemia relapse. Recent identification of pre-leukemic stem cells (pre-leukemic HSCs) in AML further elucidated the cell of origin and biological consequences of initiating events in AML. Investigation of genetic, epigenetic, and proteomic profiles in AML is summarized in other articles. This review will mainly focus on the key findings of AML LSC biology, pre-leukemic HSCs, to understand how AML is approached and progresses as a cellular hierarchy driven by the ancestral cells.
| Characteristics of LSCs | ▴Top |
Although for a long time scientists consider tumors may arise from cancer stem cells (CSCs), as multiple types of cancers feature normal tissue organization with heterogeneity in the function of malignant cells, the existence of CSCs was not formally proved until the 1990s. Using xenograft combined with fluorescence activated cell sorting to analyze different subpopulations of cells from AML patients, John Dick laboratory first isolated AML LSCs. They demonstrate that AML is organized as a hierarchy originated from a primitive LSC with self-renewal ability to repopulate leukemia in immune-deficient mice [4, 5]. Following the discovery of AML LSCs, CSCs have been found in substantial types of solid malignancies including breast, colon, brain, prostate cancers, etc., proving the CSC model in the cancer field [6, 7].
Definition of LSCs
LSCs are defined by three criteria, i.e., they can 1) populate immune-deficient mice with the xenograft that recapitulates the patient disease, 2) self-renew to reproduce and sustain the disease by serial passage in xenograft assays at clonal cell doses, and 3) re-establish leukemia heterogeneity by self-renewal LSCs, colony-forming progenitors and non-proliferative leukemia blasts. By this definition, LSCs are different from leukemia-initiating cells which are only defined by their abilities to initiate leukemia but with or without self-renewal ability. LSCs are rare primitive cells usually enriched in CD34+38- population and share multiple properties with normal HSCs: CD34+38- immunophenotype, the capacity of self-renewal, cell-cycle quiescence, and requirement of microenvironment (stem cell niche). However, the origin of LSCs is not only primitive HSCs as LSCs are also present in other populations based on CD34 and CD38 expression [8]. Studies in mouse MLL-AF9 leukemia suggest LSCs can originate either from HSCs or from myeloid progenitors, with different disease behaviors depending on cell context [9-11].
Quiescence, self-renewal and clonal evolution of LSCs
While LSCs share similar properties with normal HSC counterparts making them difficult to be eradicated from patients, substantial investigations have characterized the properties specific for LSCs (Fig. 1). LSC compartments, similar to other CSCs, are heterogeneous at self-renewal potential with enhanced self-renewal ability and evolutional properties following treatment or in vivo passages [12, 13]. Minor dormant LSC clones evolve and fluctuate, and become dominant clones to contribute to leukemia progression and relapse. MicroRNA-126 is one of the important regulators for LSC functions as Lechman et al showed that through targeting the PI3K/AKT/MTOR signaling pathway, microRNA-126 preserved quiescence, increased self-renewal and promoted chemo-resistance of LSCs. Dorrance et al showed that LSCs can be targeted in vivo using antagomiR-126 nanoparticles, resulting in LSC reduction [14]. Id2/E-protein axis was shown as another key LSC regulator to orchestrate mouse LSC self-renewal and differentiation for initiation and maintenance of MLL-rearranged AML [15]. Patients with MLL-rearranged AML who had higher Id2 gene expression had better survival than those with lower Id2 expression. In contrast, overexpression of Id2 expanded normal human HSCs, and reduction of Id2 expression induced lymphoid lineage priming, suggesting that Id2 plays a converse role in normal HSCs and LSCs [16].
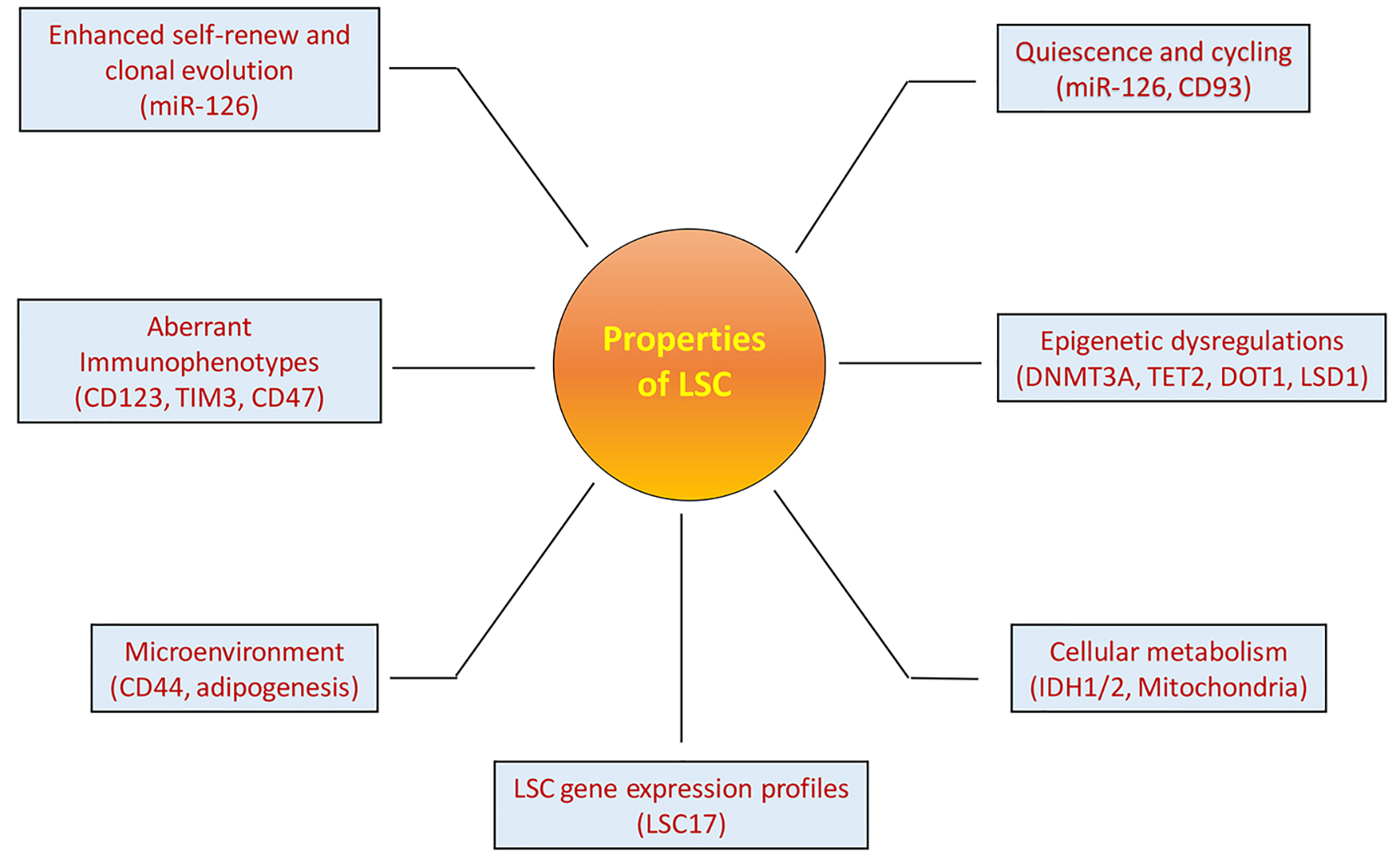 Click for large image | Figure 1. Properties of LSCs. LSCs have multiple specific properties that are important for LSCs to maintain leukemia clones and contribute to leukemia progression and relapse. These properties are also important for identification and targeting of LSCs as discussed in the article. The names in the brackets under each property are the example characteristics of LSCs. |
LSC cell-surface markers
During leukemogenesis LSCs aberrantly express certain cell-surface markers, phenotypically distinguished from HSCs. The first immunophenotype found for LSCs is the production of interleukin-3 receptor α (CD123) antigen, which is highly expressed in both leukemic blasts and CD34+38- LSC population. In contrast, CD123 is minimally expressed in the normal HSC population from adult bone marrow, making Cd123 a unique and interesting target for eradicating LSCs [17]. Multiple anti-CD123 antibody-based strategies have been developed, which include modified monoclonal antibodies, bispecific antibodies or conjugates of anti-CD123 antibody, toxins or electron-emitting radioisotope, and the creation of chimeric antigen receptor (CAR) modified T cells [18-22].
Other identified LSC cell-surface markers such as TIM3, C96, CLL-1, CD25, CD32 and CD47 [20-25], also provide interesting targets for eliminating LSCs. Increased CD47 expression on LSCs and other cancer CSCs enhances its binding to the receptor Sirpa on macrophages and induces innate immune tolerance. Disruption of CD47-Sirpa interaction induces macrophage-mediated phagocytosis of LSCs and other CSCs [25, 26]. The identification of LSC-specific antigens provides promising targets for developing antibody-based immunotherapies against LSCs, although the success of immunotherapies remains to be determined in clinical trials.
Microenvironment of LSCs
LSCs reside in endosteal microenvironment to maintain stemness, quiescence and resistance to chemotherapy. Using an antibody targeting adhesion molecule CD44 on LSCs, Jin et al demonstrated that LSCs require interactions with supportive microenvironment for self-renewal and proliferation [27]. This study, in parallel with a similar observation in CML LSCs, provided clear evidence that disruption of the supportive component of LSC niche is a new avenue to eliminate LSCs [28]. In a mouse study, gonadal adipose tissue was observed as an alternative niche for fatty acid transporter CD36+ LSCs to evade chemotherapy [29]. However, a recent study showed that AML cells compromised adipocyte bone marrow niche and leukemia growth can be repressed through therapeutically induced BM adipogenesis [30]. Collectively, these findings and other observations [31] have elucidated the importance of stem cell niche for LSC function, providing new means for developing novel therapies to target LSC stemness at the root through modulating the LSC-niche interaction.
Genetic and epigenetic dysregulations in LSCs
By genetic models, genetic lesions that drive carcinogenesis for continuous cancer growth, including substantial chromosomal translocations and gene mutations have been found in AML cases and in LSCs [32, 33]. While genetic changes and their roles in leukemic process are well documented in AML, LSCs also create LSC-specific transcription-factor-driven gene expression programs. Multiple intrinsic signaling pathways associated with stem cells such as HOX genes, Wnt/β-catenin, Hh pathways and inactivation of p53 tumor-suppressor pathways have been found to be critical for LSC survival and proliferation [34, 35].
In addition to genetic abnormalities extensively investigated in AML and LSCs, recently numerous studies have revealed the important role of epigenetic dysregulations in AML and LSCs. Most of driver mutations found in early stages of leukemogenesis are genes associated with epigenetic regulations including DNMT3A and TET2 [33, 36, 37]. An LSC DNA methylation signature derived from xenograft was shown largely mutation independent and associated with poor prognosis [38]. A recent study by Cimmino et al showed that genetic restoration of TET2 function or treatment with vitamin C blocked aberrant self-renewal of LSC in mouse TET2-deficient leukemia and suppressed leukemia progression [39]. Lysine methyltransferase DOT1L causes aberrant H3K79 methylation in MLL-rearranged leukemia. Inhibition of DOT1L selectively impairs LSCs with very limited side effects on normal hematopoiesis [40]. Lysing demethylase LSD1 is highly expressed in patients with AML and other cancers [41]. Inhibition of LSD1 demethylase reactivates the all-trans-retinoic acid differentiation pathways in AML, resulting in the regression of AML growth in xenograft with impairment of repopulating secondary mice [42]. Collectively, these studies demonstrate that both major areas of epigenetics, DNA methylation and histone modification are functionally involved in LSC function for leukemia development and progression.
| Clinical Relevance of LSCs | ▴Top |
LSCs are mostly defined in experimental xenograft assays based on their capacities of generating and maintaining malignant clones in mice. Although some studies showed LSCs may contribute to failure of treatment, metastasis and poor survival, for example, the frequency of putative AML LSCs (CD34+38-) has a strong prognostic impact on patients’ survival but the neoplastic population (CD34+38+ and CD34-) shows no prognostic impact [43, 44], the clinical relevance and importance of LSC concept has long been questioned. Using gene expression microarray analysis data generated from functionally validated LSC population of AML samples in xenograft assays, Eppert et al created an LSC signature that shares a core transcriptional program with normal HSCs and is a highly significant independent predictor of patient survival [8]. Patients with LSC gene expression signature have poorer prognosis than those without LSC profiles. This study provides the first clear evidence that the LSCs identified using the xenograft mouse model for human AML are clinically relevant. This was further confirmed by gene and epigenetic expression profiles generated from different groups [45, 46]. To develop a better predictive biomarker for identification of AML patients with high risk, Ng et al generated a list of genes related to stemness from functionally defined LSCs using the same strategy Eppert et al adopted but with a larger number of samples [47]. An optimal 17-gene LSC score (LSC17) was further generated from the list of the genes through sparse regression analysis against survival in a large training cohort. The LSC17 score was highly prognostic and predictive of initial therapy response/resistance to current treatments including allogeneic stem cell transplantation. These studies demonstrate that the LSC model in AML is not only clinical relevant but also important for clinicians to better determine the prognostic risk and evaluate novel strategies to improve the survival of patients with high risks.
| Pre-Leukemic HSCs in AML | ▴Top |
Like other malignancies AML is also considered a clonal disease growing from a single stem/progenitor cell that has sequential acquisition of fewer but certain genetic mutations than solid tumors [48, 49]. This stepwise evolutional leukemogenesis generates inter-tumor genetic diversity and cellular heterogeneity consisting of LSCs with variable self-renewal capacities, committed colony-forming AML progenitors, and non-proliferative blasts. Recently, a more ancestral pre-leukemic HSC was identified as the new source and compartment of AML heterogeneity [36, 50, 51]. Pre-leukemic HSCs must be defined as a condition that a HSC has some but not all leukemia-specific mutations that cause clonal hematopoietic expansion without disease, but are associated with progression to AML when additional leukemia-specific mutations acquired. The existence of pre-leukemic HSCs was indicated by clonal analysis over three decades ago [52, 53] and was extensively studied in pediatric twin pairs with concordant and discordant AML, evidenced by the discovery of the same genetic alterations in the twin patients or in the normal differentiated cells of the non-leukemia twin [54, 55]. Yasuda et al reported a donor-derived leukemia in patients after allo-transplant of HSCs from a HLA-matched sibling but the donor whose blood cells carried IDH2 and DNMT3A mutations remained free of leukemia 10 years after his donation, suggesting the presence of subclinical pre-leukemic hematopoiesis [56]. A recent study by Slush et al directly identified pre-leukemic HSCs in AML at diagnosis. When large-scale deep sequencing was performed on highly purified HSC progenitors and mature cells from AML patients, HSCs bearing DNMT3A mutation resulted in competitive multilineage repopulation advantage over non-mutated HSCs in xenograft. DNMT3A mutations were found present in stem/progenitor cells at diagnosis and remission, with increased allele frequency in late remission suggesting that pre-leukemic HSCs are chemo-resistant. In contrast, both DNMT3A and NPM1 mutations were widely found in all kinds of cells from relapse, demonstrating that DNMT3A mutation occurs earlier in HSCs, leading to clonal expansion of pre-leukemia before full leukemia developed by additional leukemia-specific NPM1 mutation [36]. Pre-leukemic HSCs were also found in patients with other mutations such as TET2, IDH2, IKZF1, and ASXL1 followed by secondary leukemia-specific mutations like NPM1, FLT3ITD, JAK2 and NRAS for the development of full leukemia in patients and in relapse [50, 51]. Collectively, the identification of pre-leukemic HSCs has elucidated both the cell of origin for mutations and the order of mutation acquisition in leukemogenesis toward development of leukemia (Fig. 2). Studies that examined the important role of pre-leukemic HSCs in leukemia and relapse provide broad clinical implications. In addition to LSCs, the more ancestral pre-leukemic HSCs in patients should also be monitored and targeted for the prevention of leukemia relapse.
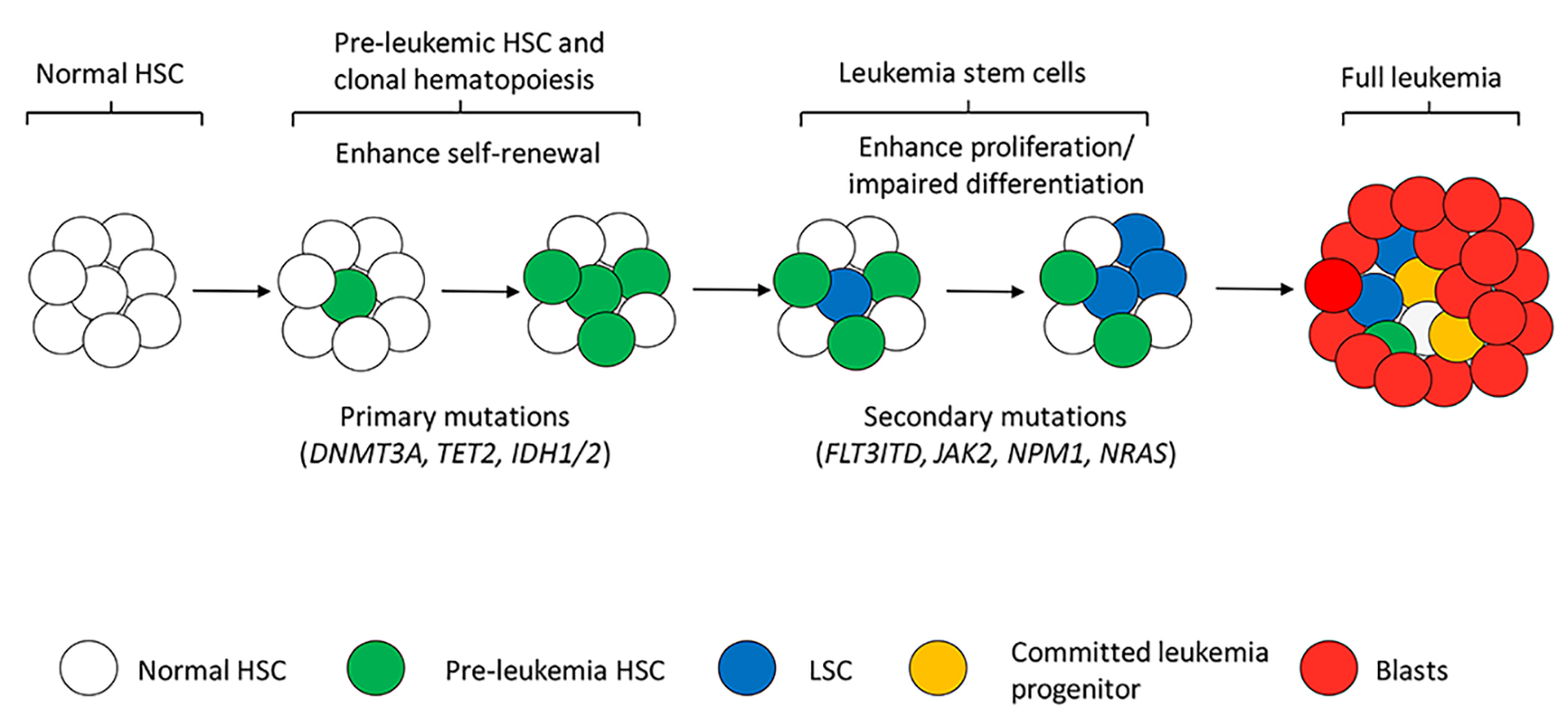 Click for large image | Figure 2. Simplified model of multistep leukemogenesis and leukemia heterogeneity. Acquisition of primary mutations mainly DNMT3A, TET2, IDH1/2 and others modifies NDA methylation status in normal HSCs, resulting in enhanced self-renewal and clonal hematopoiesis (pre-leukemic HSCs). Pre-leukemic HSCs with the primary mutations are responsible only for clonal hematopoiesis or early stage of leukemic process which is not irreversible to leukemia transformation. Acquisition of secondary mutations like FLT3-ITD and JAK2 provides pre-leukemic HSCs the ability of proliferation. Mutated HSCs with both self-renewal and proliferation abilities are considered LSCs that create leukemia clones, produce daughter progenitors and blasts cells. Together distinct types of cells form the complex leukemia heterogeneity. Differentiation programs should also be affected by the genetic alterations in LSCs for accumulation of immature blasts in frank leukemia. |
| Cell Origin of AML Relapse | ▴Top |
Despite achieving remission, majority of AML patients die from relapse. It used to be generally accepted that the mechanism for leukemia relapse was that leukemic cells acquire drug-resistant mutations or alterations, either as a consequence of the mutagenic effect of chemo-drugs or due to the intrinsic instinct of leukemia cells to generate genetic alterations for drug resistance [57, 58]. However, drug-resistant leukemia cells are recently considered to pre-exist and survive from chemotherapy as pre-leukemic HSCs, LSCs and their associated progenitors in the heterogeneous AML respond differently to chemotherapies. Studies on dynamics of genetically or lentivirus marked LSCs and CSCs showed that not all LSCs or CSCs are equal in their tumor propagation ability as functional diversity among LSCs and CSCs were detected. Some minor undetectable drug-resistant cells are selected and evolved by chemotherapy and as a consequence, expanded to become dominant clones [12, 13]. Similar to LSCs, pre-leukemic HSCs are also putative reservoirs for leukemia relapse because of their stemness properties of dormancy, self-renewal and capacity of evolution following chemotherapy with increased frequency in remission and relapse [36]. Nevertheless, it remains unclear how AML patients in remission relapse and whether pre-leukemic HSCs and LSCs directly contribute to relapse. Recently Shlush et al showed the complexity of leukemia progression between individual patients. They found that leukemia progresses to relapse by two different therapy-resistant cell types with genetic and immunophenotypic dissimilarities [59]. Relapse originates in some cases from rare LSCs that arise from pre-leukemic HSCs present in primitive HSCs/progenitors at diagnosis and repopulate in NSG mice. In another instance, relapse variants detected by variant allele frequency are not observed in any primitive HSCs/progenitors at diagnosis or in repopulated NSG mice. Those relapse variants present in CD33+ leukemia blasts at diagnosis demonstrate that this type of disease relapse originates from phenotypically more committed leukemia cells. Investigations of gene expression profile exposed both groups of disease require stem cell properties for relapse, either LSCs directly giving rise to relapse or stemness transcriptional programs being retained or regained in more matured leukemia cells. In summary, identification of pre-leukemic HSCs by Shlush et al sheds light on the mechanisms underlying AML progression and relapse (summarized in Fig. 3).
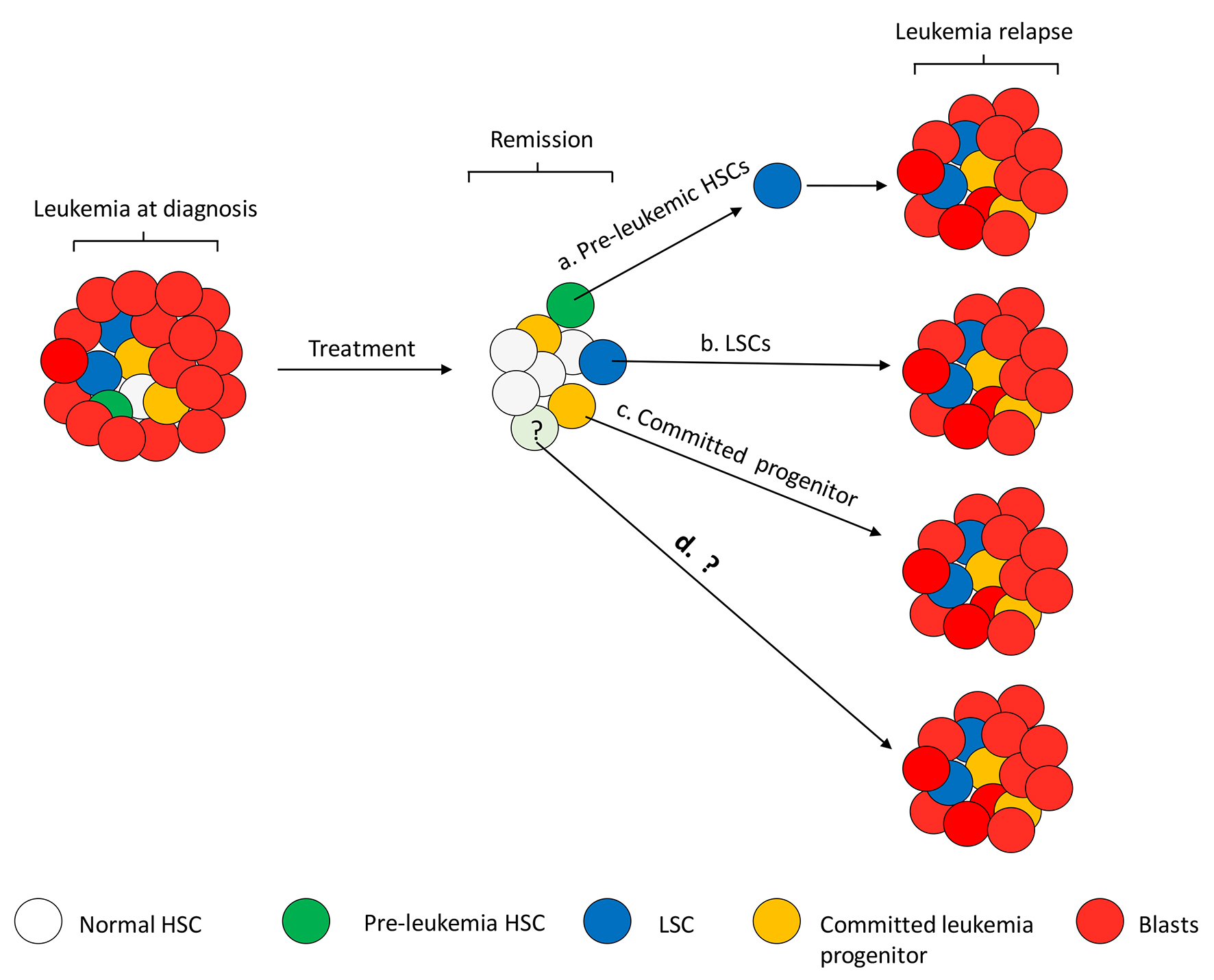 Click for large image | Figure 3. Speculated mechanism of leukemia relapse. Relapse can happen at any time in AML patients with complete remission (CR), from as early as a couple of months to a few or up to 5 years since CR is achieved. Proliferation of different types of residual cells in a patient’s BM may contribute to relapse. To summarize the studies by Shlush et al, relapse could arise from a rare pre-leukemic HSC (e.g. DNMT3A) that needs to acquire additional mutations (e.g. NPM1) to become LSCs and result in relapse (a), or rare dormant LSCs that are resistant to chemotherapies proliferate for leukemia relapse (b). Relapse from rare pre-leukemic HSCs or LSCs takes longer time than from more committed progenitor cells (c). Not all the patients tested in the studies by Shlush et al fit into the categories of cell of origin for relapse, indicating other mechanisms may be also involved in relapse (d). |
While the study by Slush et al first elucidates cell origin of relapse and provides key insight into leukemia pathophysiology, progression of leukemia to relapse could be more complicated than what we have discovered. In their study, not all patients fell into these two patterns of relapse, indicating that either a more sensitive sequencing technology needs to be adopted for detection of rarer relapse variants, or other types of cells or genetic events are responsible for leukemia relapse (Fig. 3). Also, the phenomenon of relapse from committed mature leukemic cells suggests that either founder dominant AML clones are incompletely eradicated by inductive therapies, or they inherit chemo-resistance from their ancestral stem cells at diagnosis.
| Conclusions | ▴Top |
Like other solid cancers, AML also results from multistep pathogenesis, related to cytogenetic abnormalities, point mutations, aberrant gene expression, and epigenetic dysregulations. Numerous studies on AML LSC biology have indicated that non-genetic mechanisms together with genetic and epigenetic alterations are involved in leukemogenesis and leukemia progression. This review mainly summarized recent advances of studies on LSC properties to elucidate the non-genetic machinery and their clinical implications. Identification of more ancestral pre-leukemic HSCs provides key insight into the cell origin of leukemic initiation and the order of mutation acquisition during leukemia development. Genes that are first mutated are usually the ones (e.g. DNMT3A, TET2, IDH1/2, and ASXL1) involved in epigenetic regulation or cellular metabolism in HSCs causing defect in normal hematopoiesis and clonal evolution. Additional late mutations (e.g. FLT3-ITD, NPM1, JAK2, and NRAS) are more leukemia specific and usually contribute to leukemic cell growth through constitutive activation of signaling pathways for development of frank leukemia. The recognition of persistence of LSCs and pre-leukemic HSCs in leukemia development and remission, and their contribution to relapse will prompt the development of new therapies for the eradication of these ancestral malignant cells. Multiple inhibitors have been produced to target those mutations and are in the process of clinical trial but impressive efficacies like the success of imatinib in chronic myeloid leukemia have not yet been achieved. Development of new therapeutic approaches relies on deeper insight into the pre-leukemic/leukemic stem cell properties. Recent findings of metabolic abnormalities in LSCs (e.g. IDH1/2 mutation-induced aberrant enzymatic activity and increased mitochondrial mass and fatty-acid oxidation) should offer a new promising avenue to design therapeutic strategies [60-63]. Individualized disease profiles focusing on characterization and monitoring of LSCs, pre-leukemic HSCs and other biomarkers like LSC17 for patients with AML should be generated so that clinicians can choose the optimal therapeutic options and novel strategies for the management of relapse.
| References | ▴Top |
- Dohner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015;373(12):1136-1152.
doi pubmed - Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 2005;15(9):494-501.
doi pubmed - Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275-291.
doi pubmed - Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645-648.
doi pubmed - Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730-737.
doi pubmed - Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105-111.
doi pubmed - O’Brien CA, Kreso A, Jamieson CH. Cancer stem cells and self-renewal. Clin Cancer Res. 2010;16(12):3113-3120.
doi pubmed - Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, Metzeler KH, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17(9):1086-1093.
doi pubmed - Chen W, Kumar AR, Hudson WA, Li Q, Wu B, Staggs RA, Lund EA, et al. Malignant transformation initiated by Mll-AF9: gene dosage and critical target cells. Cancer Cell. 2008;13(5):432-440.
doi pubmed pmc - Krivtsov AV, Figueroa ME, Sinha AU, Stubbs MC, Feng Z, Valk PJ, Delwel R, et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia. 2013;27(4):852-860.
doi pubmed pmc - Stavropoulou V, Kaspar S, Brault L, Sanders MA, Juge S, Morettini S, Tzankov A, et al. MLL-AF9 expression in hematopoietic stem cells drives a highly invasive AML expressing EMT-Related genes linked to poor outcome. Cancer Cell. 2016;30(1):43-58.
doi pubmed - Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5(7):738-743.
doi pubmed - Kreso A, O’Brien CA, van Galen P, Gan OI, Notta F, Brown AM, Ng K, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. 2013;339(6119):543-548.
doi pubmed - Dorrance AM, Neviani P, Ferenchak GJ, Huang X, Nicolet D, Maharry KS, Ozer HG, et al. Targeting leukemia stem cells in vivo with antagomiR-126 nanoparticles in acute myeloid leukemia. Leukemia. 2015;29(11):2143-2153.
doi - Ghisi M, Kats L, Masson F, Li J, Kratina T, Vidacs E, Gilan O, et al. Id2 and E Proteins orchestrate the initiation and maintenance of MLL-Rearranged acute myeloid leukemia. Cancer Cell. 2016;30(1):59-74.
doi pubmed - van Galen P, Kreso A, Wienholds E, Laurenti E, Eppert K, Lechman ER, Mbong N, et al. Reduced lymphoid lineage priming promotes human hematopoietic stem cell expansion. Cell Stem Cell. 2014;14(1):94-106.
doi pubmed - Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, Meyerrose T, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14(10):1777-1784.
doi pubmed - Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L, Guthridge MA, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5(1):31-42.
doi pubmed - Liu K, Zhu M, Huang Y, Wei S, Xie J, Xiao Y. CD123 and its potential clinical application in leukemias. Life Sci. 2015;122:59-64.
doi pubmed - Kikushige Y, Shima T, Takayanagi S, Urata S, Miyamoto T, Iwasaki H, Takenaka K, et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7(6):708-717.
doi pubmed - Jan M, Chao MP, Cha AC, Alizadeh AA, Gentles AJ, Weissman IL, Majeti R. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci U S A. 2011;108(12):5009-5014.
doi pubmed pmc - Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M, Krensky AM, et al. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci U S A. 2007;104(26):11008-11013.
doi pubmed pmc - van Rhenen A, van Dongen GA, Kelder A, Rombouts EJ, Feller N, Moshaver B, Stigter-van Walsum M, et al. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood. 2007;110(7):2659-2666.
doi pubmed - Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, Takagi S, Uchida N, et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci Transl Med. 2010;2(17):17ra19.
doi pubmed pmc - Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD, Jr., van Rooijen N, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138(2):286-299.
doi pubmed pmc - Theocharides AP, Jin L, Cheng PY, Prasolava TK, Malko AV, Ho JM, Poeppl AG, et al. Disruption of SIRPalpha signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J Exp Med. 2012;209(10):1883-1899.
doi pubmed pmc - Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12(10):1167-1174.
doi pubmed - Krause DS, Lazarides K, von Andrian UH, Van Etten RA. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med. 2006;12(10):1175-1180.
doi pubmed - Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, Stevens B, et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. 2016;19(1):23-37.
doi pubmed pmc - Boyd AL, Reid JC, Salci KR, Aslostovar L, Benoit YD, Shapovalova Z, Nakanishi M, et al. Acute myeloid leukaemia disrupts endogenous myelo-erythropoiesis by compromising the adipocyte bone marrow niche. Nat Cell Biol. 2017;19(11):1336-1347.
doi pubmed - Schepers K, Campbell TB, Passegue E. Normal and leukemic stem cell niches: insights and therapeutic opportunities. Cell Stem Cell. 2015;16(3):254-267.
doi pubmed pmc - Bullinger L, Dohner K, Dohner H. Genomics of acute myeloid leukemia diagnosis and pathways. J Clin Oncol. 2017;35(9):934-946.
doi pubmed - Hassan C, Afshinnekoo E, Li S, Wu S, Mason CE. Genetic and epigenetic heterogeneity and the impact on cancer relapse. Exp Hematol. 2017;54:26-30.
doi pubmed - Hu Y, Li S. Survival regulation of leukemia stem cells. Cell Mol Life Sci. 2016;73(5):1039-1050.
doi pubmed - Qi J, Singh S, Hua WK, Cai Q, Chao SW, Li L, Liu H, et al. HDAC8 Inhibition Specifically Targets Inv(16) Acute Myeloid Leukemic Stem Cells by Restoring p53 Acetylation. Cell Stem Cell. 2015;17(5):597-610.
doi pubmed pmc - Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506(7488):328-333.
doi pubmed pmc - Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, Mollica L, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44(11):1179-1181.
doi pubmed pmc - Jung N, Dai B, Gentles AJ, Majeti R, Feinberg AP. An LSC epigenetic signature is largely mutation independent and implicates the HOXA cluster in AML pathogenesis. Nat Commun. 2015;6:8489.
doi pubmed pmc - Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, Ng V, et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell. 2017;170(6):1079-1095 e1020
doi pubmed - Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20(1):66-78.
doi pubmed pmc - Hayami S, Kelly JD, Cho HS, Yoshimatsu M, Unoki M, Tsunoda T, Field HI, et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int J Cancer. 2011;128(3):574-586.
doi pubmed - Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, Klein HU, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012;18(4):605-611.
doi pubmed pmc - Terwijn M, Zeijlemaker W, Kelder A, Rutten AP, Snel AN, Scholten WJ, Pabst T, et al. Leukemic stem cell frequency: a strong biomarker for clinical outcome in acute myeloid leukemia. PLoS One. 2014;9(9):e107587.
doi pubmed pmc - Gerber JM, Zeidner JF, Morse S, Blackford AL, Perkins B, Yanagisawa B, Zhang H, et al. Association of acute myeloid leukemia’s most immature phenotype with risk groups and outcomes. Haematologica. 2016;101(5):607-616.
doi pubmed pmc - Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland MC, Heinecke A, et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008;112(10):4193-4201.
doi pubmed pmc - Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-2221.
doi pubmed pmc - Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, Arruda A, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540(7633):433-437.
doi pubmed - Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, Kempski H, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469(7330):356-361.
doi pubmed - Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059-2074.
doi pubmed pmc - Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, Majeti R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4(149):149ra118.
doi pubmed pmc - Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A. 2014;111(7):2548-2553.
doi pubmed pmc - Fialkow PJ, Singer JW, Raskind WH, Adamson JW, Jacobson RJ, Bernstein ID, Dow LW, et al. Clonal development, stem-cell differentiation, and clinical remissions in acute nonlymphocytic leukemia. N Engl J Med. 1987;317(8):468-473.
doi pubmed - Miyamoto T, Weissman IL, Akashi K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc Natl Acad Sci U S A. 2000;97(13):7521-7526.
doi pubmed pmc - Ford AM, Ridge SA, Cabrera ME, Mahmoud H, Steel CM, Chan LC, Greaves M. In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature. 1993;363(6427):358-360.
doi pubmed - Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S, Green J, et al. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science. 2008;319(5861):336-339.
doi pubmed - Yasuda T, Ueno T, Fukumura K, Yamato A, Ando M, Yamaguchi H, Soda M, et al. Leukemic evolution of donor-derived cells harboring IDH2 and DNMT3A mutations after allogeneic stem cell transplantation. Leukemia. 2014;28(2):426-428.
doi pubmed - Goldie JH, Coldman AJ. The genetic origin of drug resistance in neoplasms: implications for systemic therapy. Cancer Res. 1984;44(9):3643-3653.
pubmed - Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S. Drug resistance in cancer: an overview. Cancers (Basel). 2014;6(3):1769-1792.
doi pubmed pmc - Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, Medeiros JJF, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature. 2017;547(7661):104-108.
doi pubmed - Ragon BK, DiNardo CD. Targeting IDH1 and IDH2 mutations in acute myeloid leukemia. Curr Hematol Malig Rep. 2017;12(6):537-546.
doi pubmed - Sriskanthadevan S, Jeyaraju DV, Chung TE, Prabha S, Xu W, Skrtic M, Jhas B, et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood. 2015;125(13):2120-2130.
doi pubmed pmc - Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, Bosc C, et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017;7(7):716-735.
doi pubmed - Marlein CR, Zaitseva L, Piddock RE, Robinson SD, Edwards DR, Shafat MS, Zhou Z, et al. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood. 2017;130(14):1649-1660.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cellular and Molecular Medicine Research is published by Elmer Press Inc.